![]()
![]()
|
Partie Statistiques du cours de BioInformatiqueMaster BTV, UFR Sciences - Université d'AngersSolutions du TD numéro 1 (énoncés)
|
 Retour à la page principale de
(gH)
Retour à la page principale de
(gH)


Partie Statistiques du cours de BioInformatique
Master BTV, UFR Sciences - Université d'Angers
Solutions du TD numéro 1 (énoncés)
Les statistiques servent à décrire, synthétiser, comparer, modéliser et prédire. Pour plus de détails, consulter ma page de rappels rapides.
Une variable QT est une variable quantitative ; une QT se repère en général à ses unités et on peut additionner ses valeurs. Une variable QL est une variable qualitative ; une QL utilise en général des valeurs arbitraires (codes) non additionnables. Une variable QX est une variable textuelle ; elle peut comporter des mots, des phrases, des textes... Pour plus de détails, consulter ma page sur les variables.
Après avoir donné le nombre valeurs (ou "taille" de l'échantillon), il est d'usage de fournir les résumés nommés moyenne, variance, écart-type, coefficient de variation ainsi que médiane, quartiles, distance inter quartiles. Il est bon également d'indiquer le minimum et le maximum afin de vérifier que les données sont "raisonnables". Ces calculs ont pour but de déterminer la "tendance centrale" et la "dispersion" des données. Lorsqu'on s'intéresse à la population sous-jacente à l'échantillon, on peut aussi calculer des intervalles de confiance de la moyenne et de l'écart-type.
Avec peu de valeurs, la courbe des données a un sens. Au-delà d'une cinquantaine de valeurs, il vaut mieux utiliser un histogramme des valeurs par classe (avec les problèmes du choix du nombre et des bornes de classes que cela représente) et tracer une boite à moustache (boxplot) pour représenter la "tendance centrale" et la "dispersion" des données. On peut aussi afficher un diagramme en tige et feuilles (stemleaf) qui est semi-graphique (voir plus bas l'analyse de la variable length).
Après avoir donné le nombre valeurs (ou "taille" de l'échantillon), il est d'usage de fournir des calculs d'effectifs (ou fréquences) absolus ou relatifs, c'est-à-dire des comptages et des pourcentages. Lorsqu'on s'intéresse à la population sous-jacente à l'échantillon, on peut aussi calculer des intervalles de confiance pour certaines proportions.
Un histogramme des fréquences est souvent préférable à un diagramme circulaire sectoriel ("camembert") pour une QL car un oeil humain compare plus facilement une hauteur qu'une surface.
Excel n'est pas prévu pour gérer des colonnes mais des cellules, à l'intersection de lignes et de colonnes. Excel ne peut donc pas garantir l'homogénéité d'une colonne, ce qui est un pré-requis aux calculs statistiques.
De plus, Excel ne dispose pas de tout l'arsenal de fonctions statistiques usuelles : il ne contient que des fonctions de base. Par exemple il n'y a pas de fonction pour comparer deux moyennes, ni de graphique en boite à moustaches.
Enfin, Excel se trompe dans certains calculs. Voir la section correspondante dans ma page statgen.
Accession est un identifiant (ni QT ni QL) ; length, foldindex, pi, mw et gravy sont des QT ; reign, pfam, cdd, genre, espèce sont des QL.
Le calcul direct en R pour la longueur peut se faire avec les instructions suivantes une fois les données chargées et la variable lng définie :
# Calculs avec R pour la longueur : nbp <- length( lng ) # tendance centrale moy <- mean( lng ) med <- quantile(lng, 0.5) # dispersion dis <- var( lng ) ect <- sd( lng ) cdv <- round( 100*(ect/moy) ) qu1 <- quantile(lng, 0.25) qu3 <- quantile(lng, 0.75) diq <- qu3 - qu1 # valeurs pour controle lmi <- min( lng ) lma <- max( lng ) # affichages cat("Taille de l'échantillon ",nbp," protéines\n") cat("Moyenne ",moy," aa\n") cat("Variance ",dis," aa^2\n") cat("Ecart type ",ect," aa\n") cat("Coefficient de variation ",cdv," %\n") cat("Médiane ",med," aa\n") cat("Premier quartile ",qu1," aa\n") cat("Troisième quartile ",qu3," aa\n") cat("Distance inter quartiles ",qu3," aa\n") cat("Minimum ",lmi," aa\n") cat("Maximum ",lma," aa\n") # une fonction résumée disponible sous R summary(lng)Et on obtient alors :
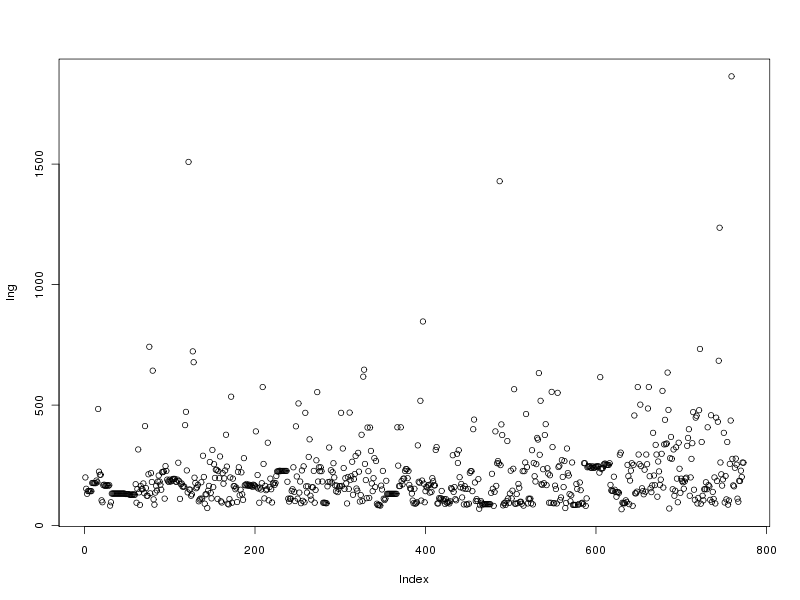
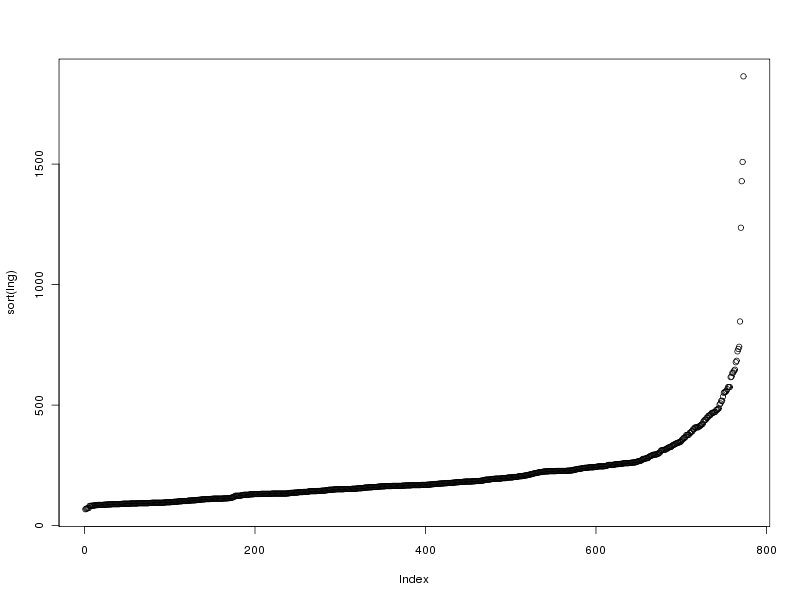
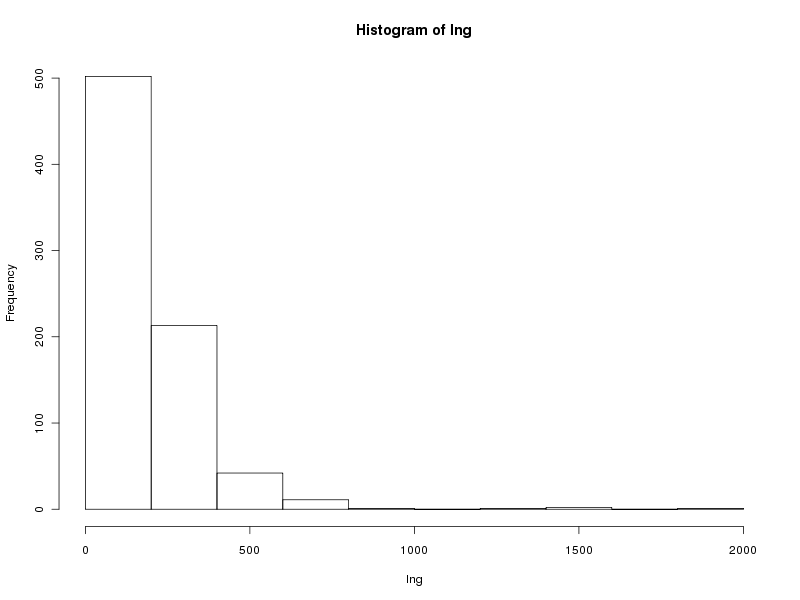
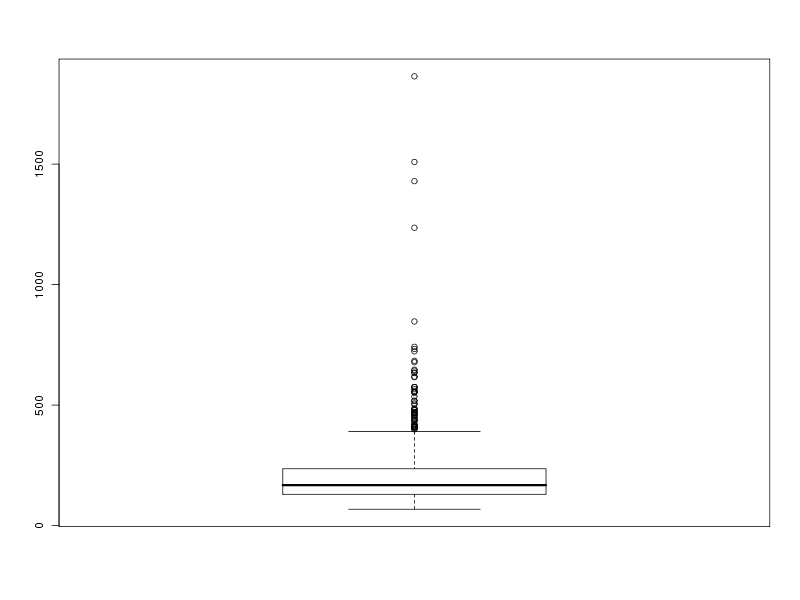
Taille de l'échantillon 773 protéines Moyenne 205.6882 aa Variance 22109.43 aa^2 Ecart type 148.6924 aa Coefficient de variation 72 % Médiane 168 aa Premier quartile 130 aa Troisième quartile 236 aa Distance inter quartiles 236 aa Minimum 68 aa Maximum 1864 aa Min. 1st Qu. Median Mean 3rd Qu. Max. 68.0 130.0 168.0 205.7 236.0 1864.0Cela fait donc beaucoup de choses à écrire, des affichages de sortie mal cadrés, alors que l'idée est simple : on veut juste décrire la variable lng. Pour les tracés associés, on peut écrire :
# tracé des valeurs plot(lng) # tracé des valeurs triées plot(sort(lng)) # histogramme des valeurs par classe (découpage automatique) hist(lng) # tracé en boite à moustache boxplot(lng)Ce qui fournit les 4 graphiques suivants (cliquables) :
A l'aide des fonctions de statgh.r, tout va beaucoup plus vite à écrire :
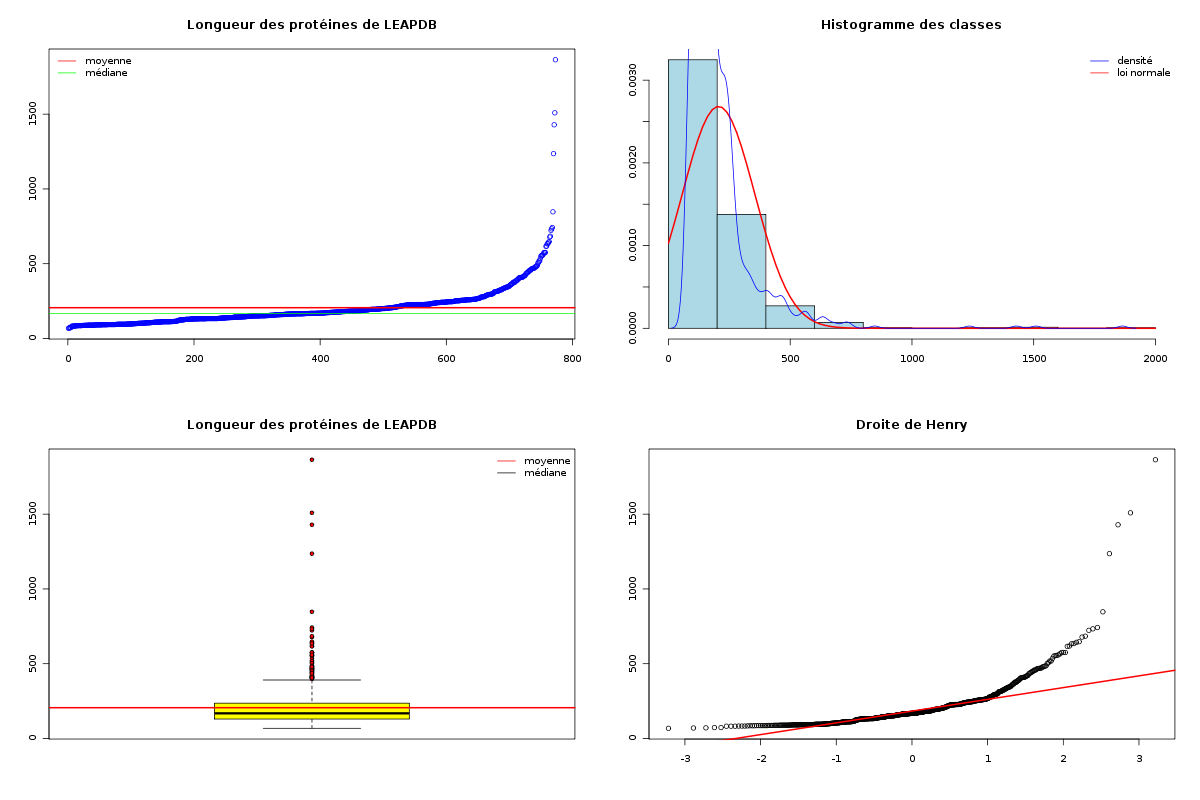
source("statgh.r") ; lea <- lit.dar("lea.dar") lng <- lea[,1] decritQT("Longueur des protéines de LEAPDB",lng,"aa",TRUE)De plus les sorties sont bien présentées, tout est écrit en français et les graphiques sont en couleurs : (sur les graphiques, la moyenne est en rouge, la médiane en vert)
VARIABLE Longueur des protéines de LEAPDB Taille 773 individus Moyenne 205.6882 aa Ecart-type 148.5962 aa Coef. de variation 72 % 1er Quartile 130 aa Mediane 168 aa 3eme Quartile 236 aa iqr 106 aa Minimum 68 aa Maximum 1864 aa Tracé tige et feuilles 0 | 77777888888899999999999999999999999999999999999999999999999999999999 1 | 00000000000000000000000000000000000000000000000111111111111111111111+323 2 | 00000000000000000000000000001111111111111111222222222222223333333333+107 3 | 000000011111222223333344444445556666788889999 4 | 0011111112223444556666777778889 5 | 0122455667888 6 | 22344588 7 | 234 8 | 5 9 | 10 | 11 | 12 | 4 13 | 14 | 3 15 | 1 16 | 17 | 18 | 6
L'étude de la variable reign est plus court à écrire en R :
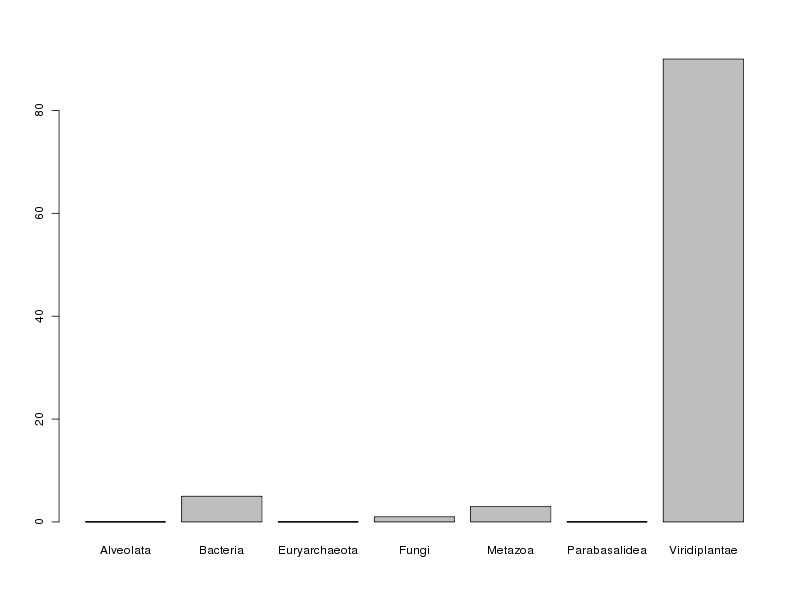
# calculs QL effs <- table(reg) tria <- round(100*table(reg)/length(reg) ) # affichage cat("Effectifs absolus\n") print( effs ) cat("Effectifs relatifs (\"tri à plat\")\n") print( tria ) # histogramme des fréquences relatives barplot(tria)et on obtient :
Effectifs absolus Alveolata Bacteria Euryarchaeota Fungi Metazoa Parabasalidea Viridiplantae 1 38 1 11 23 1 698 Effectifs relatifs ("tri à plat") Alveolata Bacteria Euryarchaeota Fungi Metazoa Parabasalidea Viridiplantae 0 5 0 1 3 0 90
A l'aide des fonctions proposées, là encore, c'est plus court :
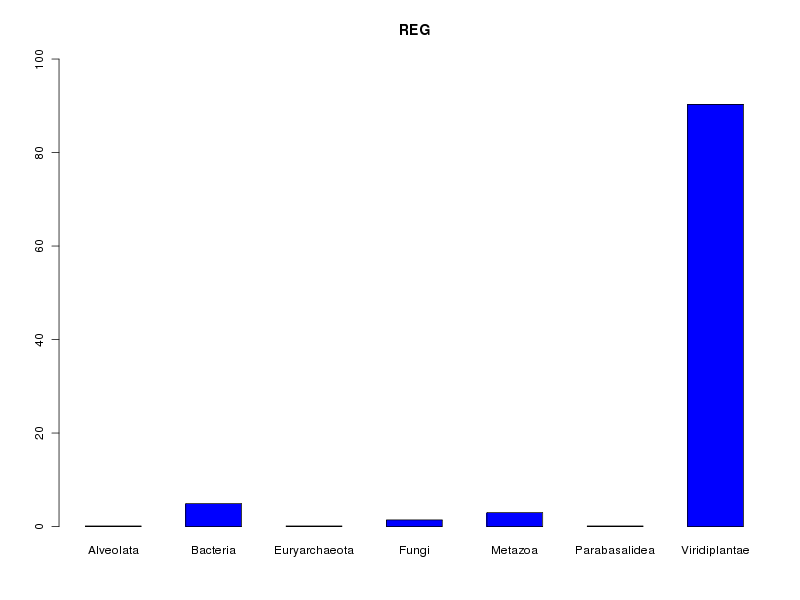
reg <- lea[,2] decritQL("REG",reg,levels(reg),TRUE)On dispose alors d'un rappel de la taille de l'échantillon et l'histogramme est dans une échelle fixe qui va de 0 à 100 % :
QUESTION : REG (ordre des modalités) Alveolata Bacteria Euryarchaeota Fungi Metazoa Parabasalidea Viridiplantae Total Effectif 1 38 1 11 23 1 698 773 Frequence (en %) 0 5 0 1 3 0 90 99 QUESTION : REG (par fréquence décroissante) Viridiplantae Bacteria Metazoa Fungi Alveolata Euryarchaeota Parabasalidea Total Effectif 698 38 23 11 1 1 1 773 Frequence (en %) 90 5 3 1 0 0 0 99
Maintenant que tous les calculs et graphiques sont faits, les statistiques commencent : il reste à rédiger ce que l'on voit dans ces calculs et graphiques.
La première partie des commentaires est scientifique et objective : elle met en phrases l'information visible dans ces résultats. On pourrait ainsi écrire : la base de données LEAPDB constitue un grand échantillon d'un peu plus de 770 protéines avec une longueur moyenne d'un peu plus de 205 aa (acides aminés) et un écart-type sur la longueur d'environ 150 aa ; les protéines présentes dans la LEAPDB ont, dans les 3/4 des cas une longueur inférieure à 236 aa.
La seconde partie des commentaires est subjective et contextuelle. Elle utilise l'expertise du domaine pour dépasser la première partie. On pourrait ainsi ajouter : ces longueurs sont donc dans une "moyenne raisonnable" de longueurs de protéines, avec toutefois quelques protéines comportant un très grand nombre d'acides aminés.
Concernant la partie subjective, on pourra remarquer que le wiki anglais parle d'une longueur moyenne de 300 aa (sans toutefois citer de source) alors que le wiki français ne dit rien à ce sujet. Un article très intéressant de Brocchieri et Karlin (Nucleic Acids Research, 2005) dont une copie locale est ici permet de mieux comprendre ces valeurs et de distinguer les longueurs des Eucaryotes et Procaryotes. On pourra également consulter les données statistiques du site UniProtKB à la rubrique 3. SEQUENCE SIZE :
http://expasy.org/sprot/relnotes/relstat.html
En ce qui concerne la variable reign, on peut dire que la très grande majorité des protéines (90 %) correspond au règne végétal (Viridiplantae) avec un petit "pool" de bactéries (5 %) et de métazoaires (3 %). Contextuellement, cela s'explique par le fait que les protéines LEA sont traditionnellement associées aux plantes.
En toute rigueur, non, pas sur l'ensemble des données car trois règnes rencontrés (Alveolata, Euryarchaeota et Parabasalidea) ne comportent qu'une seule protéine dans notre base de données ce qui n'est pas suffisant pour une étude statistique. De plus Viridiplantae est un règne très "large" pour lequel il faudrait sans doute distinguer les monocotylédones des dicotylédones mais les données fournies dans le fichier lea.dar ne permettent pas d'accèder à ce niveau de détail.
Avec l'instruction R t.test(lng) on obtient l'intervalle de confiance de la longueur. Avec nos fonctions, on peut utiliser la forme courte ou la forme longue de icmQT :
icmQT(lng) icmQT(lng,0.05,TRUE)qui fournit les résultats suivants :
> icmQT(lng) 195.1897 216.1868 > icmQT(lng,0.05,TRUE) Pour 773 valeurs de moyenne 205.6882 et d'écart-type 148.6924 au niveau 0.05 la valeur critique issue de la loi du t de Student pour 772 ddl est 1.963042 soit 1.963 l'intervalle de confiance est donc sans doute [ 195.1897 ; 216.1868 ] soit, en arrondi, [ 195.190 ; 216.187 ]En ce qui concerne la normalité, l'histogramme des valeurs par classe ne semble pas correspondre à une courbe en cloche et la distribution des données, notamment visible par le diagramme tige et feuilles l'indique également. Mais en toute rigueur, il faudrait utiliser un test statistique. On appréciera le tracé de la normale sous-jacente et le tracé automatique de la droite de Henry avec notre fonction decritQT qui confirme tout cela.
Celui de Cornilon et al. Voir la couverture sur ma page MFE. L'utilisation de Rcmdr y est également décrite.